Apakah Marfan Syndrome?
Sindrom Marfan menggambarkan gangguan keturunan yang rumit pada tisu penghubung, yang terutama memberi kesan kepada mata, sistem kardiovaskular dan sistem otot rangka. Walau bagaimanapun, memandangkan setiap organ terdiri daripada tisu penghubung, sindrom Marfan dengan idealnya boleh memusnahkan dan mengganggu dengan pertumbuhan dan fungsi setiap tapak anatomi.
Sindrom ditransmisikan sebagai sifat dominan autosomal: oleh itu kita menghadapi penyakit genetik yang serius, mempunyai ekspresi fenotip yang sangat berubah (cacat boleh berbeza dari keluarga ke keluarga atau dari pesakit ke pesakit).
Apa yang mencetuskan sindrom Marfan adalah perubahan gen FBN1 (di kromosom 15), yang kod untuk fibrillin-1, glikoprotein penyambung yang sangat penting yang merupakan sokongan struktur untuk mikrofibrils.
Microfibrils: yang terdiri daripada fibrillin, mikrofibrils hadir dalam matriks ekstraselular, di mana mereka membentuk web untuk pemendapan elastin dalam gentian anjal. Walaupun terdapat ubiquitous di dalam badan, mikrofibrils banyak terdapat di aorta, ligamen dan zonul badan-badan ciliary (di tingkat ocular).
Menjadi penyakit dominan autosomal, hanya kanak-kanak yang mewarisi gen FBN-1 yang diubah oleh kedua ibu bapa yang terjejas oleh sindrom Marfan. Walau bagaimanapun, dalam satu kes daripada 4 penyakit itu adalah hasil mutasi spontan pada pesakit yang tidak mempunyai sejarah keluarga.
Nama penyakit itu berasal dari ahli pediatrik Perancis yang pertama kali menyifatkannya pada tahun 1896 (A. Marfan), selepas itu ia harus menunggu sehingga tahun 1991 untuk mengenal pasti gen yang diubah terlibat dalam manifestasi gejala: penemu itu adalah F. Ramirez.
Tonton Video
X Tonton video di youtubepunca
Kami telah menyebutkan bahawa Sindrom Marfan adalah ekspresi segera dari mutasi gen yang kod untuk fibrillin-1. Marilah kita cuba untuk memperdalam topik ini untuk memahami mekanisme yang ditetapkan untuk mencetuskan sindrom.
FIBRILLINA 1 adalah komponen elastin glikoprotein, penting untuk memastikan dan mengekalkan keanjalan dan kekuatan tisu. Dalam keadaan fisiologi, fibrillin 1 mengikat protein lain, yang dikenali sebagai TGF-beta (atau mengubah faktor beta pertumbuhan). TGF-beta nampaknya terlibat dalam proses-proses yang merosakkan yang mempengaruhi otot licin vaskular dan matriks ekstraselular. Bermula dari asumsi-asumsi ini, beberapa penulis yakin bahawa Sindrom Marfan adalah disebabkan, sebagai tambahan kepada mutasi gen FBN-1, juga kepada lebihan TGF-beta, terutama di aorta, di dalam injap jantung dan di dalam paru-paru.
kejadian
Adalah dianggarkan bahawa Sindrom Marfan memberi kesan kepada 1 subjek setiap 3, 000-5, 000 kelahiran dan memperlihatkan dirinya secara tidak jelas antara lelaki dan perempuan, tanpa predileksi untuk baka yang tepat. Statistik menunjukkan bahawa 75% pesakit mempunyai sejarah keluarga yang positif; dalam baki 25% penyebabnya terletak pada mutasi sporadis yang nampaknya dikaitkan, dalam beberapa cara, dengan usia lanjut ayah pada masa pembuahan.
Kanak-kanak yang mempunyai sindrom Marfan yang teruk mempunyai jangka hayat kurang dari satu tahun.
Sebelum evolusi strategi pembedahan jantung terbuka, kebanyakan pesakit dengan sindrom Marfan mempunyai jangka hayat purata 32 tahun; terima kasih kepada peningkatan terapi terapi perubatan dan farmakologi, kini pesakit Sindrom Marfan hidup secara purata sehingga 60 tahun.
Tanda dan gejala
Untuk mengetahui lebih lanjut: Gejala Sindrom Marfan
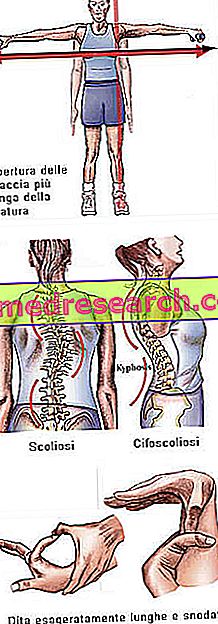
Marfan sindrom boleh berjalan sepenuhnya tanpa gejala. Pesakit yang terjejas mempunyai struktur yang lebih langsing, yang tidak seimbang dan nipis. Bahagian bawah dan atas mempunyai panjang lebih tinggi daripada batang (dolicostenomegalia). Terdapat juga perbincangan arachnodactyly untuk mengungkapkan konsep panjang jari-jari yang dibesar-besarkan, yang tipikal dari subjek yang dipengaruhi oleh Marfan syndrome: tangannya berbanding dengan kaki labah-labah.
Bagi ketinggian, pesakit ini mempunyai ketinggian purata di atas persentil ke-97.
Ciri khas lain yang sering dijumpai pada pesakit dengan sindrom Marfan termasuk:
- Lengan terbuka lebih besar daripada ketinggian
- Sendi longgar → mobiliti bersama dibesar-besarkan
- Pengubahsuaian dinding dada
- Anjakan kristal
- Bahagian atas badan kurang maju daripada bahagian bawah
- Pneumothorax spontan (11%)
- scoliosis
- Striae kulit pada paha, belakang, deltoid, tahap dada
Di antara tanda-tanda yang paling bermasalah yang berkaitan dengan sindrom Marfan, kami menyebutkan prolaps injap jantung dan kekurangan injap mitral: keadaan yang sama dengan mudah dapat mempercepat pelebaran cincin aorta dan pembedahan aorta.
Jadual menunjukkan tanda-tanda yang boleh dijumpai pada pesakit dengan sindrom Marfan. Watak-watak yang diterangkan tidak selalu ada, tetapi bahagian yang baik dapat dijumpai.
| Laman anatomi terjejas | Gejala yang mungkin |
Cute | Striae di kawasan toraks, lumbar dan sakral |
mata | Kemandian visi, astigmatisme, detasmen retina, glaukoma sudut sempit, lilitan lensa, miopia |
Struktur tulang | Arthralgia, kyphoscoliosis, dolicostenomelia (perkembangan yang berlebihan dalam panjang anggota badan yang berkaitan dengan batang), hipermobilitas, lelangit yang tinggi, dada yang cacat, kaki rata, pergelangan tangan yang sempit dan nipis, bantahan masuk semula / sternum yang tidak normal, scoliosis, bahu melengkung, spondylolisthesis |
Dita | arachnodactyly |
paru-paru | Pneumothorax spontan, dyspnea, penyakit obstruktif pulmonari idiopatik |
Perubahan wajah | Ogivalate palate (malformation of the palate), retrognathia mandibular (cacat dalam perkembangan rahang), muka panjang |
hati | Angina pectoris, aneurisma aortic abdomen, arrhythmia jantung, pembesaran aorta toraks / pecah / dissection, kekurangan aorta, prolaps injap mitral |
bahasa | Kesukaran bahasa |
diagnosis
Memandangkan lebih daripada 200 mutasi mungkin, penggunaan penanda genetik hampir mustahil untuk tujuan diagnostik.
Penentuan Sindrom Marfan tidak selalu begitu segera, memandangkan ekspresi fenotip mutasi tidak selalu jelas dan mudah dikenalpasti. Kelewatan diagnostik secara serius boleh menjejaskan kelangsungan hidup pesakit: hanya berfikir, sebagai contoh, kegagalan untuk mengenali masalah kardiovaskular.
Kriteria diagnostik bagi Marfan syndrome telah dibuat di peringkat antarabangsa pada tahun 1996: diagnosisnya terdiri dari penyiasatan sejarah keluarga yang dikaitkan dengan gabungan indikator utama dan kecil sindrom.
Beberapa ujian diagnostik yang digunakan ialah:
- ekokardiogram
- Angiorisonance magnetik dan CT (untuk penyiasatan aorta)
- angiografi resonans magnetik (MRA) dengan cecair kontras (untuk membuat struktur dalaman aorta jelas)
- pemeriksaan dengan lampu celah (untuk menganalisis kehalusan lensa yang mungkin)
- pengukuran tekanan okular (untuk menyerlahkan kemungkinan kehadiran glaukoma)
- ujian genetik (disyorkan sebelum mengandung kanak-kanak untuk menentukan sindrom atau tidak)
terapi
Menjadi penyakit genetik, tidak ada ubat atau rawatan yang boleh membalikkan penyakit itu.
Walau bagaimanapun penggunaan ubat-ubatan adalah penting untuk mengurangkan gejala-gejala dan mengelakkan sebarang komplikasi, terutama pada jantung. Untuk tujuan ini, ubat-ubatan untuk mengurangkan tekanan darah, seperti sartans (terutamanya), inhibitor ACE dan beta-blocker, sangat sesuai.
Dalam konteks Sindrom Marfan, pesakit juga menderita scoliosis boleh mengikuti penjagaan khusus, serta bagi mereka yang terkena glaukoma.
Pembedahan boleh dibetulkan untuk membetulkan dilarutkan aorta, satu elemen yang sering menyatukan sebahagian besar pesakit dengan sindrom Marfan.
Teruskan: Marfan Syndrome - Dadah dan Penjagaan »


