keluasan
Sindrom Noonan adalah penyakit kelainan genetik yang kadang kala keturunan, yang mengubah perkembangan normal pelbagai bahagian anatomi badan. Ciri-ciri wajah yang luar biasa, kedudukan pendek dan beberapa kecacatan jantung kongenital adalah tanda-tanda patologi dan diagnostik utama.

Akibat patologi tidak selalu dramatik: dalam sesetengah kes, sebenarnya, sindrom Noonan dapat menyebabkan simptomologi bernuansa dan membolehkan kehidupan normal.
Peringatan genetik ringkas: DNA dan kromosom
Sebelum menerangkan sindrom Noonan, adalah baik untuk membuat rujukan ringkas kepada genetik.
CHROMOSOMES DAN DNA
Setiap sel manusia sihat mempunyai 23 pasang homolog kromosom : 23 adalah ibu, iaitu, warisan dari ibu, dan 23 adalah ayah, atau warisan dari bapa. Sepasang kromosom ini adalah seksual, iaitu, ia menentukan jantina individu; baki 22 pasang, sebaliknya, terdiri daripada kromosom autosomal . Keseluruhannya, 46 kromosom manusia mengandungi keseluruhan bahan genetik, lebih dikenali sebagai DNA . Dalam DNA seorang individu ditulis ciri-ciri somatiknya, kecenderungannya, kebolehan fizikalnya dan sebagainya.
GENES DAN MUTASI DNA
DNA diorganisasikan dalam pelbagai urutan, lebih kurang panjang, dipanggil gen . Setiap gen menduduki kromosom tertentu dan rakan sejawatannya, kerana ia terdapat dalam dua salinan, yang disebut alel . Alel berasal dari ibu dan tinggal di kromosom ibu; alel lain berasal dari bapa dan ditempatkan di kromosom bapa.

Rajah: penubuhan gen dalam sepasang kromosom homolog. Sepasang kromosom homolog mengandungi gen tertentu, semuanya mempunyai dua varian, alel, yang menduduki kedudukan kromosom yang sama dan melakukan fungsi yang sama (kecuali mutasi). Pasangan kiri kromosom mempunyai dua alel sama (kedua-dua biru); pasangan yang betul bukannya mempunyai dua alel berbeza (satu merah, yang lain berwarna biru).
Daripada gen protein berasal, hadir di dalam badan kita. Apabila mutasi DNA berlaku, gen (biasanya alel) dari kromosom tertentu boleh rosak dan oleh itu menghasilkan protein yang rosak.
Apa itu sindrom Noonan
Sindrom Noonan adalah sejenis penyakit genetik, kadang-kadang keturunan yang menyebabkan beberapa anomali anatomi (dan bukan sahaja) di beberapa bahagian badan. Perubahan utama berlaku di muka, jantung dan sistem tulang, tetapi mungkin sistem pembiakan, sistem limfatik, kulit, sistem saraf, mata, telinga, darah dan buah pinggang.
Keabnormalan yang mencirikan sindrom Noonan tidak selalu sama dalam semua pesakit: dalam sesetengah mereka kabur, sehingga mereka hampir tidak disedari dan membolehkan kehidupan yang hampir normal; Dalam orang lain, sebaliknya, mereka sangat menonjol untuk membahayakan mereka yang memakainya.
Epidemiologi
Sindrom Noonan tidak biasa: sebenarnya, ia memberi kesan kepada seorang kanak-kanak setiap 2, 500 bayi yang baru lahir. Lelaki dan perempuan sama-sama terjejas dan tidak ada satu etnik yang lebih terdedah daripada yang lain.
punca
Sindrom Noonan adalah penyakit genetik, oleh itu ia disebabkan oleh perubahan dalam DNA . Para penyelidik yakin bahawa sekurang-kurangnya lapan gen terlibat, tetapi mereka masih tidak dapat menjelaskan dengan jelas bagaimana mereka menyebabkan keabnormalan anatomi.
Apa yang dilakukan oleh lapan gen ini?
PERANAN
Gen-gen yang terlibat dalam sindrom Noonan mempunyai peranan fisiologi tertentu: mereka menghasilkan protein yang membolehkan pertumbuhan dan perkembangan sel-sel kita. Dengan kata lain, protein ini adalah hujung pengawalseliaan, yang menjaga kehidupan dan takdir setiap sel tunggal dengan terperinci.
Apabila gen yang mencipta protein ini bermutasi, mekanisme pengawalan selular yang diterangkan di atas juga diubah, dengan kerosakan yang serius kepada tubuh.
THE GENESE INVOLVED
Daripada lapan gen yang terlibat dalam sindrom Noonan, hanya empat yang telah dikenal pasti sepenuhnya, kerana mereka berubah dengan frekuensi yang agak ketara.
Mereka adalah:
- PTPN11 : mutasinya mencirikan sekitar 50% kes. Ia berada pada kromosom 12 dan penemuan implikasinya dalam sindrom Noonan bermula pada tahun 2001. Protein yang dihasilkannya mempunyai peranan asas semasa perkembangan embrio: ia menguasai, sebenarnya, pertumbuhan, pembezaan dan pembahagian sel, hati khususnya. Kami tahu dengan pasti bahawa alel mutasi sudah cukup untuk penyakit itu muncul (dominasi).
- SOS1 : bermutasi dalam 10-15% kes. Difahamkan dia boleh terlibat dalam penyakit itu pada tahun 2006.
- RAF1 : bermutasi dalam 5-10% kes. Peranannya tidak diketahui sehingga tahun 2007.
- KRAS : mutasinya mencirikan kira-kira 2% kes. Penemuan implikasinya adalah terkini: 2006.
Seperti yang anda dapat lihat, penyelidikan yang berkaitan dengan gen ini agak baru dan ini menjelaskan mengapa mereka masih belum selesai.
PEMULIHAN?: YA ATAU TIDAK?
Mutasi genetik, dalam 50% kes, adalah sporadis (iaitu disebabkan oleh rawak), manakala dalam baki 50% mereka disebarkan oleh salah seorang ibu bapa .
Biasanya, orang tua yang menghantar penyakit kepada anak-anak mereka tidak tahu bahawa mereka dipengaruhi oleh sindrom Noonan, kerana ini mungkin bukan bentuk yang serius dan jelas.
gejala
Sindrom Noonan memanifestasikan dirinya dengan beberapa tanda ciri dalam beberapa bahagian badan.
Tanda-tanda ini terdiri daripada anomali anatomi yang lebih atau kurang ketara dan dalam keadaan patologi yang lebih atau kurang serius. Malah, sesetengah pesakit menunjukkan penyakit ini dengan jelas, dalam setiap aspek; Sebaliknya, yang lain pula lebih nuanced atau terhad kepada beberapa laman web.
KARAKTERISTIK PATOLOGIK UTAMA DAN SEKOLAH
Ciri-ciri wajah yang luar biasa, kedudukan pendek dan serangkaian kecacatan jantung kongenital adalah kelainan yang terdapat pada semua orang yang menderita sindrom Noonan; Atas sebab ini, ciri-ciri utama penyakit itu dipertimbangkan (NB: istilah kongenital bermaksud hadir sejak lahir ).
Sebaliknya, anomali berikut dianggap sebagai ciri sekunder, kerana ia kurang biasa daripada yang sebelumnya:
- Penampilan mudah hematomas dan pendarahan
- Kesukaran pembelajaran dan anomali tingkah laku
- Kekurangan visual pelbagai jenis
- lymphedema
- Hipotonia otot
- Kehilangan pendengaran
- Kemandulan dan alat kelamin abnormal
- Makan yang sukar (pada masa kanak-kanak)
- Anomali rangka
RAWATAN TANAH
Ciri-ciri muka yang tidak normal dapat dilihat ketika pesakit sangat kecil; Dari masa ke masa, variasi mereka diperhatikan, menjadi kekal pada masa dewasa.
- Bayi yang sangat awal (kurang daripada setahun): mata jauh dari satu sama lain dan cenderung ke arah bawah. Telinga adalah rendah dan berorientasikan ke arah belakang kepala. Alur, yang ada di atas bibir atas, adalah dalam; lehernya pendek dan garis rambut di belakang kepalanya rendah.
- Masa kanak-kanak : mata mula menjadi menonjol, sementara kelopak mata diturunkan ( ptosis ) dan menjadi lebih tebal. Hidung dihancurkan pada akar, tetapi mempunyai pangkal yang luas dan ujung bulat.
- Masa kanak-kanak : ciri-ciri terdahulu ditambah ketekalan wajah, bibir yang diperbesar dan panjang muka yang lebih besar.
- Masa remaja : dahi menjadi luas dan dagu ditunjuk, sehingga menjadikan muka sama dengan segitiga. Beberapa ciri-ciri wajah yang menonjol, seperti garis yang bermula dari hidung dan mencapai sudut mulut, sementara mata menjadi kurang menonjol. Leher pendek diperkaya dengan butiran lain: lipatan pertama ( pterigium colli ) muncul dan otot trapezius lebih besar.
- Umur dewasa : alur yang berlari dari hidung ke sisi mulut adalah mendalam dan jelas. Mulut menjadi lebih menonjol dan kulit menjadi berkerut dan jelas. Mata sentiasa jauh dan dengan kelopak mata menebal dan terjejas oleh ptosis.
STATUS RENDAH
Berat dan panjang semasa kelahiran adalah normal. Walau bagaimanapun, dalam tempoh 18-24 bulan pertama, pertumbuhan diperhatikan perlahan dan tertangguh berbanding dengan kanak-kanak yang sama umur.

Rajah: wajah orang dewasa dengan sindrom Noonan. Dari laman web ini: login.aafp.org
Ini disebabkan oleh masalah hormon ( hormon pertumbuhan, GH ), tetapi juga kepada beberapa kesukaran yang dimakan oleh kanak-kanak.
Kemerosotan pertumbuhan juga diperhatikan semasa zaman kanak-kanak dan, di atas semua, semasa baligh, apabila kanak-kanak lelaki cenderung berkembang secara tiba-tiba. Perbezaan dari rakan sebaya pada masa ini dalam kehidupan jelas.
Dalam ketiadaan rawatan, ketinggian lelaki mencapai purata 162 cm, manakala pada wanita ia mencapai 153 cm. Dengan rawatan terapeutik yang tepat, ketinggian juga boleh berlaku dalam purata normal.
CONGERITAL CARDIAC DEFECTS
80% pesakit dengan sindrom Noonan dilahirkan dengan kecacatan jantung yang lebih kurang atau kurang. Anomali ini boleh:
- Stenosis injap pulmonari : ini adalah penyempitan (stenosis) injap jantung (yang membolehkan darah mengalir dari ventrikel kanan jantung ke arah arteri pulmonari). Arteri pulmonari bertanggungjawab untuk mengangkut darah ke paru-paru akibat oksigenasinya.
- Stenosis arteri pulmonari : ia adalah penyempitan arteri pulmonari; kecacatan ini, seperti sebelumnya, menghadkan jumlah darah yang mencapai paru-paru untuk oksigenasi.
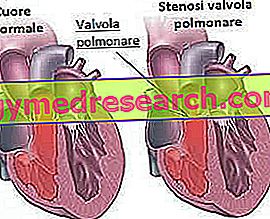
- Kekurangan septum antara ventrikel (antara ventrikel) atau antara atrial (di antara atria): terdiri daripada pembentukan lubang antara dinding tisu yang memisahkan ventrikel (atau yang memisahkan atria). Ini menyebabkan darah mengalir di antara dua petak, menyebabkan masalah peredaran darah, kadang-kadang lebih serius.

ANOMALIS LAIN
Oleh kerana ini adalah pelbagai peristiwa, kami akan cuba menggambarkannya dalam ciri utama mereka.
- Kesukaran pembelajaran dan anomali tingkah laku . Perisikan boleh lebih rendah daripada purata. Walau bagaimanapun, terdapat banyak individu yang, walaupun menjadi pembawa penyakit ini, mempunyai IQ yang normal.
Begitu juga untuk tingkah laku: dalam sesetengah kes, pesakit itu mudah marah, mengulangi bunyi dan perkataan orang lain dan mempunyai keperluan diet yang aneh.
- Penampilan mudah hematomas dan pendarahan . Dalam 50% pesakit, kebolehan pembekuan dikurangkan. Oleh itu, kehilangan darah, walaupun selepas trauma remeh, adalah mudah dilihat. Ia adalah ciri yang tidak boleh diabaikan apabila mengambil ubat tertentu (aspirin) atau menjalani pembedahan (semua, dari yang paling invasif kepada pengekstrakan gigi yang mudah).
- Kekurangan visual pelbagai jenis . Kira-kira separuh daripada pesakit mempunyai masalah penglihatan dalam pelbagai jenis. Strabismus, astigmatisme, miopia, mata malas ( amblyopia ), hipermetropia dan nystagmus adalah cacat visual yang mungkin timbul pada pesakit dengan sindrom Noonan.
- Lymphedema . Ini adalah kecacatan dalam sistem limfa . Cecair limfatik, atau limfa, berkumpul di beberapa kawasan badan, seperti tangan dan kaki. Ia berlaku terutamanya semasa zaman kanak-kanak.
- Hipotonia otot . Ini adalah pengurangan nada otot. Ia adalah tipikal zaman kanak-kanak dan zaman kanak-kanak.
- Kehilangan pendengaran . Ini adalah episod sementara semasa belia. Ia disebabkan oleh otitis yang kerap di telinga tengah.
- Kemandulan dan alat kelamin abnormal . Gangguan genital diperhatikan dalam 60% pesakit. Seringkali, subjek adalah lelaki, yang mengalami cryptorchidism (iaitu kegagalan satu atau kedua-dua buah zakar untuk turun ke skrotum). Untuk menyelesaikan masalah ini, pembedahan mesti digunakan. Jika tidak, itu tanpa campur tangan, individu itu mempunyai bilangan spermatozoa yang berkurang, oleh itu dia kurang subur.
Wanita dengan sindrom Noonan biasanya tidak mempunyai masalah ini.
- Pemakanan yang sukar . Ini adalah masalah biasa zaman kanak-kanak dan yang menyumbang untuk menangguhkan pertumbuhan. Kanak-kanak mempunyai masalah dengan sedutan susu dan cenderung muntah selepas makan.
- Anomali rangka . Terutama pada remaja muda, hipermobiliti artikular (iaitu sendi dengan pelbagai gerakan), scoliosis, dada excavatum (atau corong), dada dapat dilihat dan puting yang dipisahkan dengan cara yang luar biasa.
KETIKA DAN BAGI APA-APA YANG DAPAT DAPAT DAPAT DENGAN DOKTOR?
Sekiranya anda perasan ciri-ciri wajah seperti yang dinyatakan di atas dalam kanak-kanak, adalah dinasihatkan supaya pesakit kecil menjalani pemeriksaan pediatrik. Doktor kemudian akan membuat penilaian yang sesuai dan, jika dia mengesyaki sindrom Noonan, akan menjalankan ujian diagnostik yang diperlukan.
Dalam sesetengah kes, anomali anatomi adalah kabur, supaya mereka yang pembawa penyakit tidak tahu ia terjejas. Walau bagaimanapun, walaupun dalam keadaan ini diagnosis adalah penting, kerana masalah jantung yang mungkin timbul.
komplikasi
Komplikasi yang berkaitan dengan sindrom Noonan bergantung kepada tahap keterukan penyakit. Oleh itu, dari sudut pandangan ini, setiap pesakit mewakili kes itu sendiri.
Antara komplikasi utama, mereka sepatutnya petikan:
- Kelewatan intelektual yang serius
- Kapasiti pembekuan darah yang sangat terjejas
- Pembentukan lymphedema di sekeliling organ penting, seperti jantung dan paru-paru
- Anomali anatomi serius alat alat kelamin, dengan kesan ke atas kencing (khususnya buah pinggang)
diagnosis
Penyakit seperti sindrom Noonan, yang dicirikan oleh sejumlah besar tanda-tanda tertentu, boleh didiagnosis walaupun selepas pemeriksaan objektif . Walau bagaimanapun, dalam sesetengah kes, penyakit itu tidak begitu jelas, sehingga seperti yang telah dikatakan pada beberapa kali, ia tidak dapat disedari sehingga akil baligh atau dewasa (apabila keperluan seksual pertama pesakit timbul).
Dalam sebarang kes, untuk menjelaskan apa-apa keraguan, terdapat ujian genetik yang sangat boleh dipercayai, seperti yang disebut karyotype .
Apabila penyakit itu didiagnosis, adalah penting untuk memantau, melalui ujian tertentu, keadaan patologi yang berpotensi berbahaya bagi pesakit, seperti kecacatan jantung atau kebolehan pembekuan berkurangan. Bergantung pada hasil ujian ini, keterukan setiap kes yang dipertimbangkan boleh ditubuhkan dengan tepat.
Tanda-tanda yang paling penting dalam peperiksaan fizikal:
| Pemantauan dilaksanakan oleh:
|
rawatan
Menjadi penyakit genetik, sindrom Noonan tidak dapat disembuhkan.
Walau bagaimanapun, gejala yang dihasilkannya boleh dibatasi dan dikurangkan melalui terapi yang agak berkesan.
PENJAGAAN MASALAH HATI
Mengetahui yang mana gangguan yang menimpa jantung adalah penting untuk dapat menetapkan terapi yang paling sesuai.
Anomali jantung tertentu boleh dikawal dengan rawatan farmakologi yang mudah (diuretik, anti radikal dan beta-blocker); yang lain yang lebih serius (contohnya stenosis injap paru atau kecacatan septum) mungkin memerlukan pembedahan.
Walau bagaimanapun, pemantauan berkala adalah penting, sebagai keadaan yang tidak berbahaya dapat berubah menjadi sesuatu yang sangat serius dan perkembangan dramatik.
PENJAGAAN HORMONAL UNTUK PERTUMBUHAN
Selalunya, pembangunan semulajadi yang berkurang disebabkan oleh pengeluaran hormon pertumbuhan yang lemah, atau GH . Oleh itu, pentadbiran eksogen salah satu daripada analognya ( somatropin ), yang dihasilkan di makmal, mempunyai kesan yang sangat baik, dengan syarat program terapeutik diikuti dengan teliti. Yang terakhir adalah sangat mudah: suntikan harian, bermula dari 3-5 tahun hidup.
Kesan sampingan jarang berlaku dan kebanyakannya terdiri daripada gatal-gatal dan kemerahan di kawasan suntikan.
Pemantauan berkala tahap hormon adalah disyorkan.
PENGELUARAN DEFICIT PEMBELAJARAN
Dalam sesetengah kes, sindrom Noonan boleh menjejaskan fakulti intelektual. Dalam situasi ini, pesakit memerlukan sokongan yang sah, terutama di sekolah.
RAWATAN LAIN
Untuk kecacatan visual, kacamata yang sesuai untuk patologi yang didiagnosis mungkin cukup; jarang sekali perlu menjalani pembedahan.
Bagi masalah pembekuan, terdapat ubat-ubatan yang menggalakkan pembekuan, yang akan ditadbir pada masa keperluan. Tambahan pula, cadangan yang perlu diingatkan adalah tidak mengambil antikoagulan, seperti aspirin dan derivatifnya.
Sekiranya lymphedema dibentuk di sekitar organ kritikal seperti jantung atau paru-paru, cecair limfatik boleh disalirkan dengan memasukkan tiub khas. Pembedahan, dalam kes ini, adalah satu kemungkinan yang jarang berlaku.
Akhirnya, mengenai ketidaksuburan dan kecacatan genital, kemungkinan untuk menyelesaikan criptorchidism dengan intervensi tertentu (NB: pembaca diingatkan bahawa ketidaksuburan, dalam Noonan syndrome, adalah masalah lelaki biasa).
prognosis
Prognosis berbeza dari pesakit ke pesakit. Pada sesetengah individu, sebenarnya, sindrom Noonan menyebabkan simptomologi yang bernuansa dan membolehkan kehidupan hampir normal (dengan syarat rawatan yang sesuai digunakan); Walau bagaimanapun, dalam hal lain, ia mengompromikan dengan serius keupayaan intelektual dan kesihatan umum.
Oleh itu, prognosis boleh positif (kes pertama), tetapi juga negatif (kes kedua).
Di samping kedua-dua keadaan yang melampau ini, kes-kes yang sederhana dimasukkan, iaitu untuk mengatakan di tengah-tengah antara bentuk yang tidak serius dan yang teruk. Menghadapi keadaan ini, untuk prognosis menjadi positif, diagnosis awal adalah penting diikuti dengan rawatan yang mencukupi dan awal. Hanya boleh prognosis itu mempunyai kesan positif.
PENCEGAHAN
Malangnya, mencegah mutasi sporadis tidak mungkin. Bentuk keturunan, di sisi lain, dapat dicegah dengan memberitahu orang yang subur, menderita sindrom Noonan, kemungkinan penyebaran penyakit tersebut kepada anak-anak mereka.


